

Světová média obletěla zpráva o nové naději pro pacienty s Huntingtonovou chorobou. Vědci objevili první genovou terapii zpomalující průběh nemoci. V článcích ale často nebyl zmíněn přínos vědců z liběchovského Ústavu živočišné fyziologie a genetiky AV ČR, kteří ve vývoji léku sehráli klíčovou roli. Právě zde totiž probíhaly preklinické testy na prasatech, které řídili Jan Motlík a Zdeňka Ellederová, s níž přinášíme rozhovor.
Stojíte spolu s holandskou firmou UniQure za světovým úspěchem – potenciálním lékem na Huntingtonovu chorobu. Málokterému vědci se přitom povede vidět reálné dopady svého výzkumu na život lidí. Jaký je to pocit?
Je to bezvadné. V tomto máme v aplikovaném výzkumu výhodu oproti základnímu výzkumu, který končí publikacemi. My totiž můžeme sledovat, kam výsledky schované ve vědeckých publikacích opravdu mohou vést. Jsme o trochu blíž klinice.
Původně jsem chtěla být lékařkou, ale maturitní ročník jsem studovala v USA, kde jsem neměla biologii a chemii, a tak jsem bohužel po návratu neudělala přijímací zkoušky na medicínu a šla místo toho na VŠCHT. Někdy mě to mrzí, protože mám ráda práci s lidmi a myslím, že bych snad byla dobrou lékařkou. Takhle ale zase mohu pacientům pomoct jiným způsobem.
Jak jste se dostala k výzkumu Huntingtonovy choroby na liběchovských miniprasatech?
Po studiu biomedicínského inženýrství jsem do Liběchova přišla dělat doktorát v oblasti reprodukce prasat, zabývala jsem se regulací vzniku (neboli translace) proteinů. Dříve totiž bylo hlavním posláním liběchovského ústavu zkoumat reprodukci zvířat, dělat základní výzkum samičích pohlavních buněk (oocytů) a raně embryonálního vývoje, nebo dělat krevní testy pro šlechtění nových plemen.
Pak jsme odjeli s manželem na osm let do USA na postdoktorandský pobyt, do San Diega a do Pensylvánie. Když jsem se vrátila, tak už v Liběchovském ústavu mezitím z iniciativy Jana Motlíka vzniklo v roce 2012 Centrum PIGMOD – Pig Models of Diseases, kde se studovaly některé choroby na prasečích modelech: Huntingtonova choroba, poškození míchy anebo lidské melanomy. Byly tu vybudované moderně vybavené laboratoře a operační sál. Cílem centra bylo hledat biomarkery těchto onemocnění a zkoušet nové léčebné postupy. Dělali jsme tu třeba i transplantace kmenových buněk do míchy.
Měla jste k Huntingtonově chorobě nějaký vztah?
Naštěstí nikdo z rodiny tuto chorobu nemá. Někteří mí kolegové se takto k výzkumu dostali. Chtějí pomoct příbuzným.
Jezdíme však pravidelně na setkání pacientů s Huntingtonovou chorobou. Když jsem viděla jejich trápení, dodávalo mi to do práce velkou motivaci, abychom jim pomohli.
Pacientské organizace nám navíc na výzkum přispívaly i finančně a k takovým penězům pak přistupujete s obrovskou pokorou a snažíte se, aby byly opravdu dobře využity. Pacienti financují výzkum z vlastních peněz, aby další generace nemusely trpět jako oni. K nám vědcům, ale i k našim prasátkům, mají velký respekt. Jedna pacientka nám udělala dokonce obrázek prasete jako superhrdiny, který jsme pak měli na konferenčních taškách. Je pro mě opravdu dojemné vidět, že náš výzkum nakonec k něčemu je.
Chyba v jednom genu, která změní zdravého třicátníka v nemohoucího pacienta
V čem spočívá Huntingtonova choroba?
Je to dědičné onemocnění, které způsobuje chyba v jediném genu na čtvrtém lidském chromozomu. Ta vede k produkci mutovaného proteinu huntingtinu, jenž se pak hromadí v buňkách a zabíjí neurony. Jakmile má člověk vadný gen, vždy se u něj choroba projeví.
V tomto dotčeném genu pojmenovaném HTT se nachází úsek DNA tvořený opakováním nukleotidů CAG (cytosinu–adeninu–guaninu). Zdravý člověk má do čtyřiceti repetic CAG. Jakmile má někdo víc než 40 repetic, rozvine se u něj postupně choroba, obvykle mezi 30. a 50. rokem života. Čím víc repetic člověk má, tím dříve a silněji se choroba projeví.
Proč protein huntingtin v těle máme, když může být potenciálně tak nebezpečný?
Huntingtin produkují všichni živočichové s nervovou soustavou, od mušky octomilky až po člověka, v těle je bezesporu důležitý – reguluje mimo jiné buněčnou smrt, tzv. apoptózu. Víme, že huntingtin je nepostradatelný v embryonálním vývoji, je důležitý pro neurony, pro regulaci genů i pro transport v buňce. Bez huntingtinu se organismus nenarodí, je nepostradatelný pro vývoj.
Problém nastává tehdy, když je jedna kopie genu pro huntingtin mutovaná, tedy produkuje kromě zdravého také příliš dlouhý a špatně poskládaný protein. Později v průběhu života se tento špatný protein začne hromadit ve formě shluků v mozku, kde kvůli tomu odumírají neurony, hlavně v mozkové kůře a bazálních gangliích, což jsou oblasti odpovědné za pohyb, myšlení a emoce.
Jak se Huntingtonova choroba klinicky projevuje?
Začíná to obvykle psychickými změnami – depresemi, úzkostmi. Pak se přidávají pohybové projevy. Typické jsou mimovolné, trhavé pohyby a škubání, podle kterých se této chorobě dříve říkalo „tanec svatého Víta” neboli chorea. Sedíte s nemocným u stolu a najednou mu třeba cukne ruka a hodí po vás párek.
Nemoc dále postupuje demencí, poruchami řeči, ztrátou soběstačnosti, poruchami polykání a nakonec většinou po 15–20 letech končí úmrtím v důsledku srdečních poruch či zápalu plic způsobeného vdechnutím jídla. Často ale také život pacientů končí sebevraždou.
To je krutý osud. Kolik lidí potká?
V České republice má toto onemocnění zhruba 1 000 lidí, v celém světě možná až 800 000. Přesný počet ale neznáme.
Lze nemoc testovat a činit nějaká preventivní opatření, aby nepropukla?
Lze ji testovat prenatálně. Je pak na rodičích, zda chtějí podstoupit potrat, nebo ne. Jakmile se ale dítě narodí, tak se do 18 let nesmí testovat, aby případné potvrzení choroby neovlivnilo psychiku dítěte či dokonce nespáchalo sebevraždu.
Mnoho párů, které ví, že jeden z nich má Huntingtonovu chorobu, volí radši umělé oplodnění. Pokud bude schválena genová terapie, na jejímž vývoji jsme se podíleli, tak se možná vše změní a choroba se bude diagnostikovat co nejdříve, aby mohla být léčba včas podána.
Jak vznikla prasnice Adéla s Huntingtonovou chorobou
Mohou mít zvířata také Huntingtonovu chorobu?
To nevíme, zatím to nikdo nepopsal. Ale víme, že téměř všichni živočichové huntingtin produkují.
Jak jste tedy vytvořili miniprase s Huntingtonovou chorobou?
Injikovali jsme do jednobuněčných prasečích embryí lentivirus s lidskou mutovanou DNA sekvencí. Potom jsme laparoskopicky embrya vložili do dělohy prasnice, která nám porodila geneticky modifikované selátko.
To byla ještě stará, vlastně trochu náhodná metoda – nevěděli jsme, zda se sekvence správně zařadí do genomu a nenaruší nějakou kódující sekvenci, ale měli jsme štěstí a fungovalo to. V roce 2009 se nám narodilo prasátko Adéla, zakladatelka našeho chovu transgenních liběchovských miniprasat s Huntingtonovou chorobou, generace F1. Dnes už máme, myslím, pátou generaci jejích potomků, tedy generaci F5.
Kdybyste vytvářeli prasečí model dnes, mohli byste místo lentivirů použít molekulární nůžky? CRISPR?
Ano, dnes už je možné vytvořit i tzv. knock-in prasečí modely, kdy se pomocí nové technologie tzv. molekulárních nůžek CRISPR-Cas9 přesně najde konkrétní gen, přestřihne se a vloží se do něj žádaná sekvence, třeba mutovaný gen pro huntingtin. I takto připravená prasata u nás v Liběchově chováme.
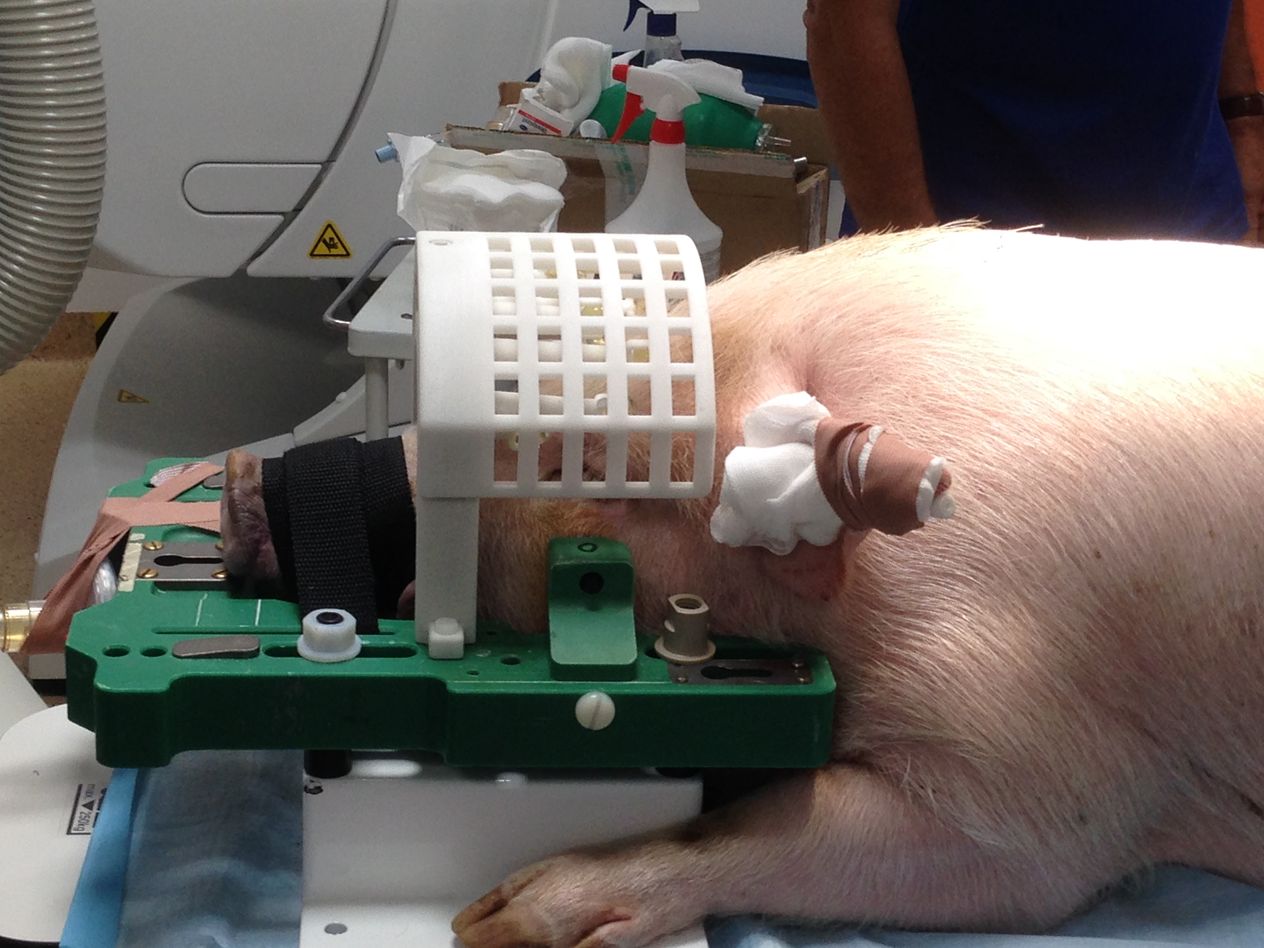
Jak na prasatech poznáte, že Huntingtonovu chorobu skutečně mají?
Kromě molekulárně genetické analýzy jsme vytvořili i sadu behaviorálních testů. Vypozorovali jsme několik mírných změn v motorice a chování, ale až v pozdním věku: prase s Huntingtonovou chorobou bylo více bojácné, bálo se jít na kladinu, vytáčelo trochu nohy při chůzi a bylo slabé v zadních nohách, takže když jsme do něj strčili, nohy mu ujely.
Spolupráce s firmou UniQure a cesta ke genové terapii
Jak jste se dostali ke spolupráci s firmou uniQure, která vedla až k vývoji genové léčby Huntingtonovy choroby?
Byli jsme od počátku v kontaktu s americkou Nadací pro Huntingtonovu chorobu, jejímž cílem bylo najít co nejdříve léčbu. Nadace nám pomohla zafinancovat vytvoření prasečího modelu.
Na jedné nadační konferenci oslovila Jana Motlíka holandská firma uniQure s tím, že vytvořili léčbu, která funguje na myších, a potřebovali by ji otestovat na prasatech. Mechanismus účinku byl postaven na RNA interferenci – tedy, že se sice vytvoří z DNA mRNA kódující huntingtin, ale pak se vlivem přípravku rozštěpí a nevznikne z ní protein.
Jak tuto léčbu dostali do buněk, kde se protein vytváří?
Používali k tomu jako vektor adeno-asociovaný virus. Na prasatech jsme ale nejdřív museli ověřit biodistribuci přípravku. Aplikovali jsme tedy virový vektor s fluorescenční svíticí značkou, abychom pak v mozku viděli, kam se virus dostal. V roce 2015 jsme začali pracovat na prvních studiích.
Jak to prakticky probíhalo?
Kolega Štefan Juhás aplikoval virus s fluorescenčním proteinem prasátkům do mozku, konkrétně do oblasti striata v bazálních gangliích, která jsou odpovědná za koordinaci pohybu. Pak jsme prasata utratili a pozorovali mozkové řezy, abychom viděli, kam všude se virus se značkou rozšířil. Rozšířil se velmi dobře, aniž by došlo k poškození buněk, jenom k mírnému zánětu vlivem fluorescenční značky.
Ověřili jste tedy správnou distribuci přípravku v mozku. Jak jste ověřili její léčebný potenciál?
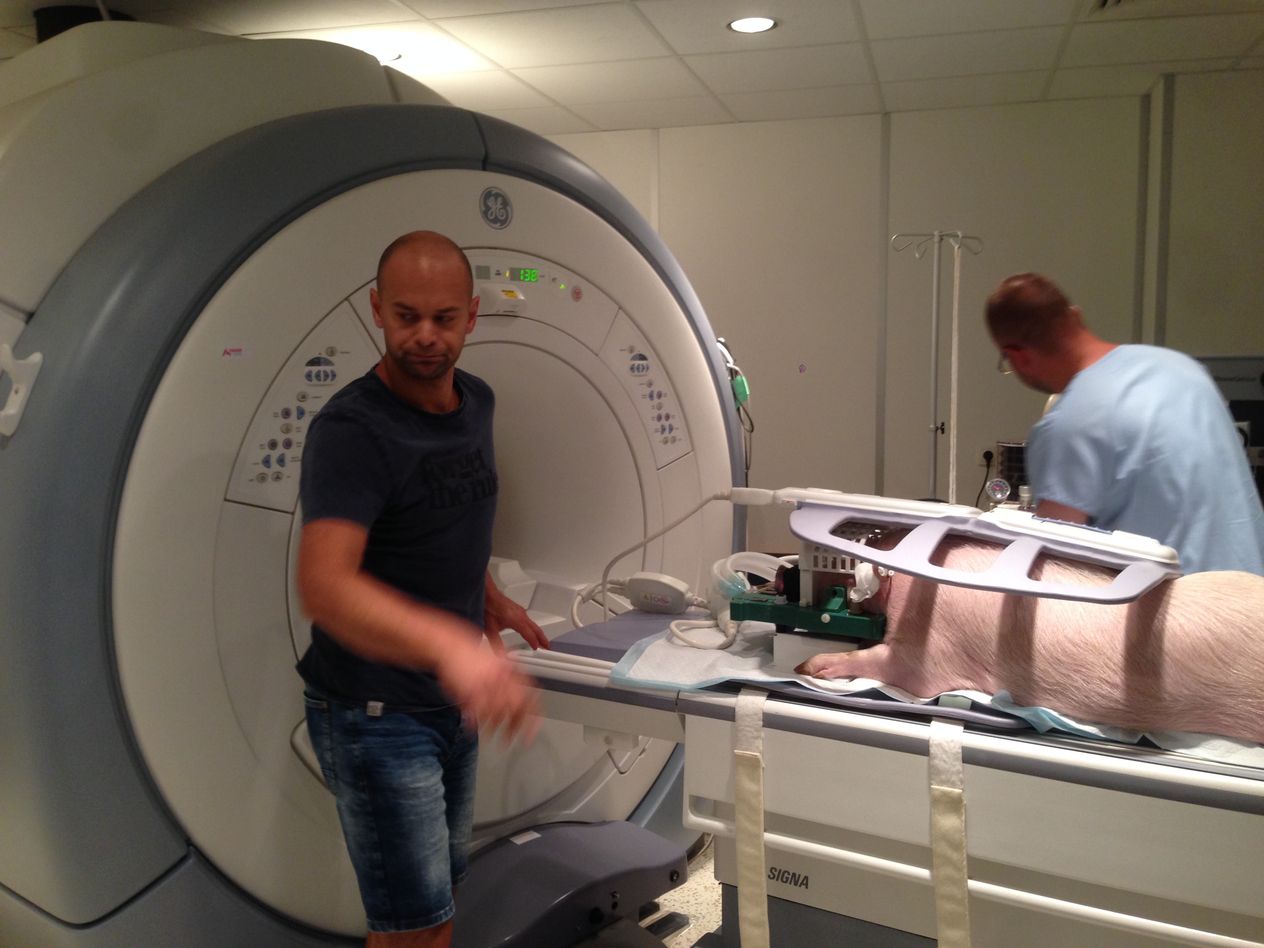
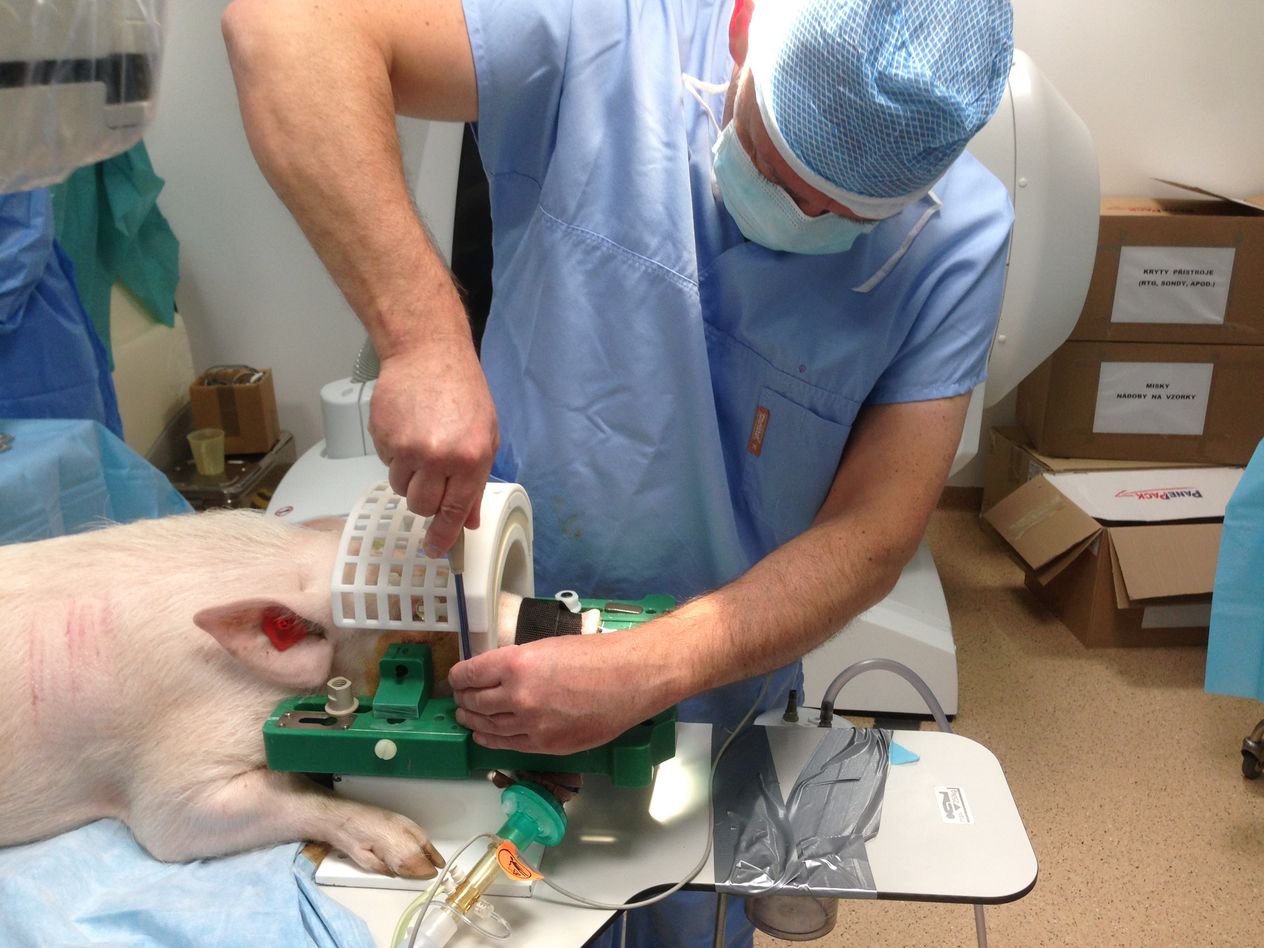
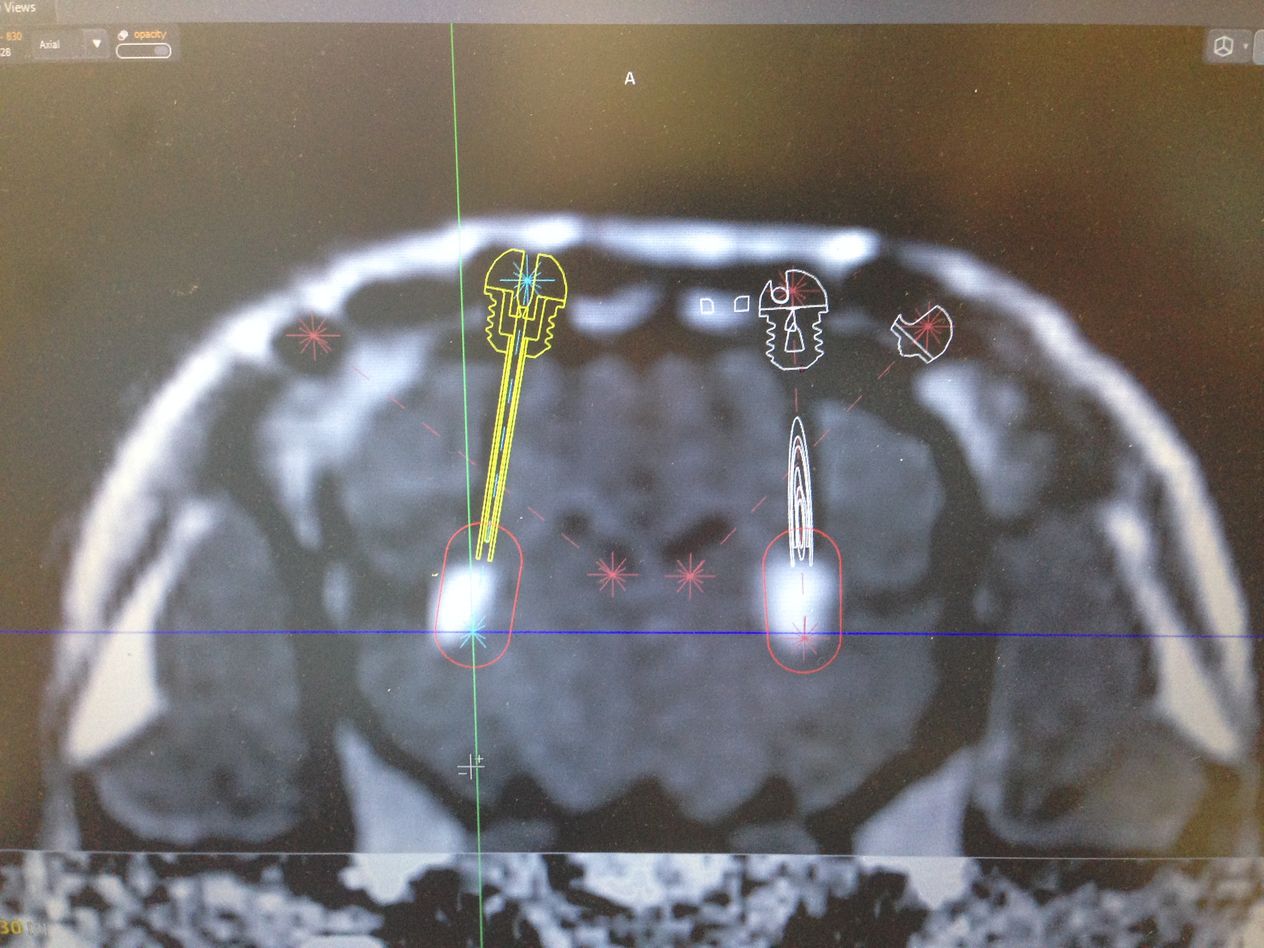
Přistoupili jsme k druhé studii, kdy už byl do viru vložen léčebný přípravek s miRNA (micro RNA), později pojmenovaný jako přípravek AMT130, a sledovali jeho účinnost. Na základě pozitivních výsledků, tedy výrazného snížení lidského huntingtinu jsme v roce 2017 spustili sérii testů na dvaceti prasatech, kdy jsme látku aplikovali do mozku kontrolovaně pomocí magnetické rezonance. Za pomoci stereotaktického nástavce a skenu z magnetické rezonance jsme navrhli koordináty, které pak použili neurochirurgové z Nemocnice na Homolce Roman Liščák a Dušan Urgošík pro navrtání malé dírky do mozku a umístění katetrů, kterými se látka pomalu do mozku aplikovala.
Jaké testy jste pak prováděli?
Sledovali jsme celkový zdravotní stav prasat a cytokinovou odpověď v krvi, abychom vyloučili zánět. Kromě krve se také prasatům odebíral pravidelně mozkomíšní mok a v obou tekutinách se sledovala hladina lidského huntingtinu a neurofilamenty, které se do tělních tekutin vylučují z mrtvých neuronů. U pacientů s Huntingtonovou chorobou je množství neurofilament v moku i krvi zvýšené.
Po půl roce jsme utratili šest prasat. My jsme pak v Liběchově udělali histologické a imunohistochemické barvení mozku, abychom zjistili, zda nedošlo k jeho poškození. Ostatní analýzy, tedy stanovení virové DNA k určení rozšíření vektoru a měření mRNA a proteinu pro ověření snížení produkce lidského mutovaného huntingtinu, již prováděla firma uniQure.
Po roce jsme utratili dalších šest prasat a společně zjistili, že efekt je ještě lepší. Tyto výsledky na velkém zvířecím modelu mohla pak firma uniQure použít jako podklady pro americký Úřad pro kontrolu potravin a léčiv (FDA), aby lék mohl být schválen pro klinické testování.
Teď máme ještě osm žijících prasat, kterým byla látka podána v roce 2017 jako malým, čtyřměsíčním prasátkům. Efekt léčby u nich stále trvá. Firma uniQure chce prokázat, že efekt je v podstatě doživotní.
Proběhly již klinické testy na lidech?
Tyto testy stále ještě probíhají. 24. 9. 2025 zveřejnila firma uniQure pozitivní výsledky z fáze I/II s AMT-130 terapií. Po třech letech u pacientů s vysokou dávkou došlo k zpomalení progrese onemocnění o 75 %. Je to první klinická studie, která prokázala zpomalení zhoršování příznaků u Huntingtonovy nemoci. UniQure plánuje podat žádost o schválení léčby v prvním čtvrtletí 2026, teoreticky by pak na konci roku 2026 mohla být komerčně dostupná. Bude však bohužel velmi drahá, jako zatím každá genová terapie a nevím, zda ji budou hradit pojišťovny.
Neexistovala by nějaká levnější metoda než navrtávat lidem mozek?
Překonat hematoencefalickou bariéru je problematické. Zkoušeli jsme ve spolupráci s uniQure látku podávat i tzv. intratekálně do mozkomíšního moku, ale to nebylo tak efektivní. Některé jiné firmy ale zkouší vyvinout adeno-asociované viry, které by prošly i hematoencefalickou bariéru, a byly takto přímo účinné v mozku.

Výzkum nám jde, ale nemáme kapacity na papírování
Obrací se na vás teď další farmaceutické firmy?
Úspěch s Huntingtonovou chorobou je poměrně čerstvý, ještě se tedy úplně nerozšířila informace o našem přínosu. Možná, že se na nás pak někdo další obrátí. Spoluprací ale máme dost.
Zaměřujeme se také na onemocnění oční sítnice, máme model pro Usherův syndrom, který je spojen s hluchotou a slepotou. Teď jsme na výzkum dostali grant od americké nadace Foundation Fighting Blindness. Máme tu také prasátko se Stargardtovou chorobou, které jsme vytvořili s německými výzkumníky z Giessenu. Kromě genové terapie zkoušíme i buněčnou terapii pro věkem podmíněnou makulární degeneraci. Na toto téma jsme v září podali se spolupracovníky z Norska grant, vypsaný dánskou farmaceutickou firmou Novo Nordisk.
Chováme 500 prasat a v tuto chvíli nemáme kapacitu na další rozšiřování, chceme se držet toho, co umíme. Chov je totiž velmi finančně náročný. Rok chovu prasat vyjde na sedm milionů a tři čtvrtiny nákladů si musíme pokrýt z projektů – grantů a smluvního výzkumu.
Takže ze mě se stala spíš manažerka než vědkyně – pořád něco píšu a sháním peníze. Ale myslím, že ta práce má smysl, a i mě to většinou baví. A také se tato manažerská práce dá lépe skloubit s rodinou. Máme s manželem, taktéž vědcem, čtyři děti.
Ve vědní politice se teď neustále skloňuje slovo transfer. Vy jste vlastně jeho zářným příkladem. Jste spokojeni s tím, jak se vám daří?
Myslím, že se máme čím chlubit. Výzkumně jsme výborní, jen asi trochu pokulháváme ve finančních otázkách, v transferu, duševním vlastnictví apod. To se ještě učíme a nemáme lidi na papírování, které je s tím spojené. Měli bychom si třeba požádat o certifikát dobré laboratorní praxe – GLP, Good Laboratory Practice. Některé studie certifikát GLP vyžadují. My ho sice reálně splňujeme, ale nemáme čas na s tím spojenou administrativu.
Zdeňka Ellederová je biochemička a od roku 2017 je vedoucí Laboratoře buněčné regenerace a plasticity na Ústavu živočišné fyziologie a genetiky AV ČR v Liběchově. Se svým týmem studuje lidská onemocnění na modelech miniprasat, sleduje jejich vývoj a testuje nové léčebné přístupy, zejména buněčnou a genovou terapii.
- Autor článku: ano
- Zdroj: VědaVýzkum.cz
{kind=link}
{kind=link}